ALS: Frühe Krankheitsmechanismen entdeckt
FAU-Forschende identifizieren Eiweiß mit pathologischen Eigenschaften
Nach aktuellem Stand der Medizin ist die Amyotrophe Lateralsklerose (ALS) nicht heilbar. Das könnte sich jedoch ändern: Forschende der FAU und der University of California San Diego (UCSD) in Kalifornien haben ein Eiweiß identifiziert, dass bereits im Frühstadium der Nervenkrankheit pathologische Eigenschaften zeigt. Die Entdeckung, die einen neuen Therapieansatz begründen könnte, hat das Team im renommierten Fachjournal „Acta Neuropathologica“ publiziert.
Im Sommer 2014 bekam die Amyotrophe Lateralsklerose, kurz ALS, durch eine Social-Media-Aktion große Aufmerksamkeit: Bei der Ice Bucket Challenge schütteten sich Millionen Menschen weltweit einen Eimer voll Eiswasser über den Kopf, um durch die Kälte das Gefühl einer Lähmung nachempfinden zu können. In Deutschland leben 6.000 bis 8.000 Menschen mit ALS, bei etwa 2.000 Patientinnen und Patienten wird die Erkrankung, die innerhalb weniger Jahre zum Tod führt, jährlich neu diagnostiziert. „ALS ist eine Motoneuronerkrankung, das heißt, es werden Nervenzellen geschädigt, die die Muskeln des Menschen steuern”, erklärt Prof. Dr. Beate Winner. „In der ersten Phase kommt es zu Muskelschwäche, später zu Muskelschwund und am Ende können die Betroffenen häufig nicht mehr schlucken und selbstständig atmen.” Mit der Aktion wurden Spendengelder gesammelt, um die Erforschung der ALS voranzutreiben.
Mit der Umwandlung in Stammzellen wird die biologische Uhr zurückgedreht
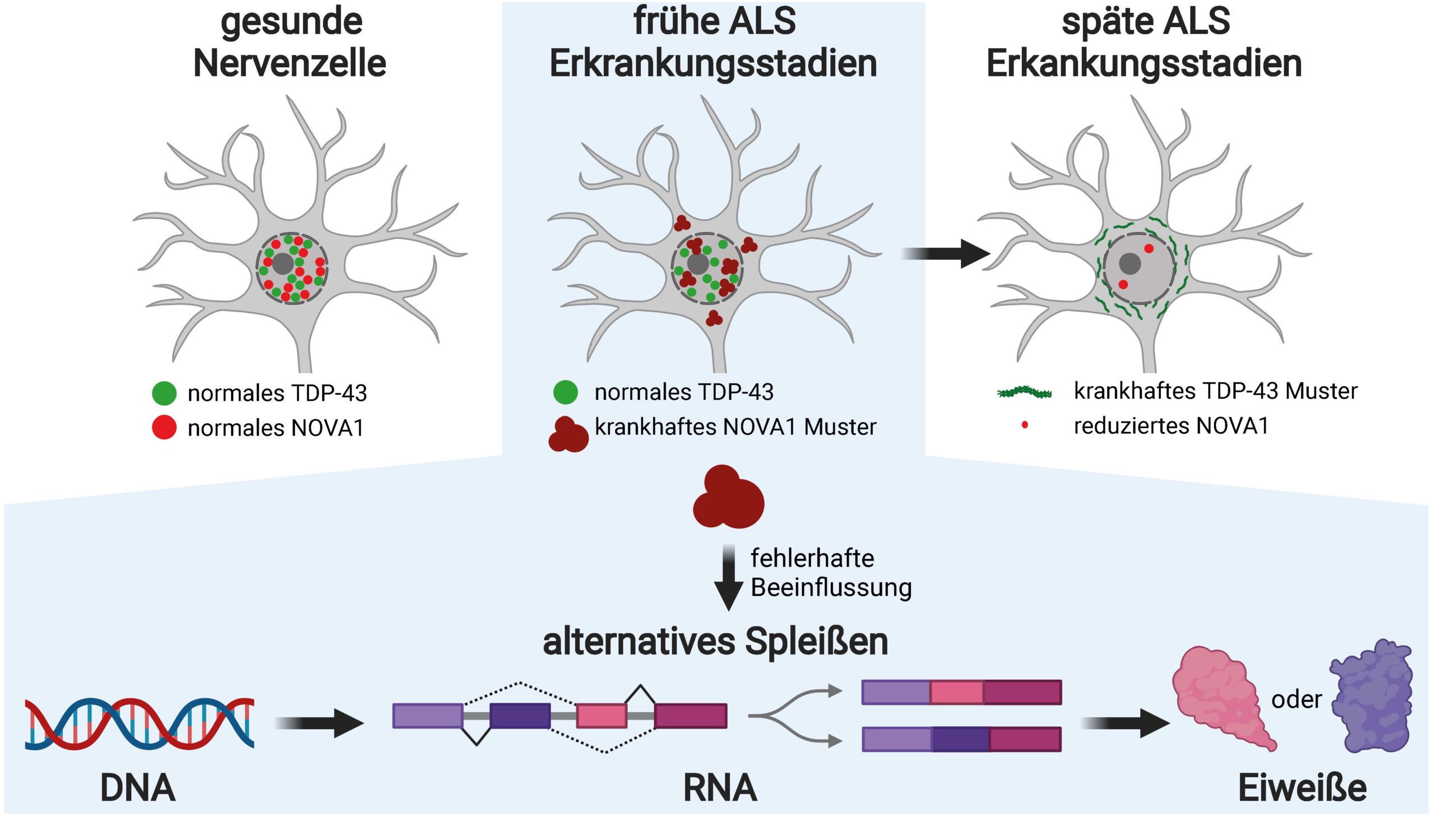
Beate Winner ist Professorin für Stammzellmodelle Seltener neuraler Erkrankungen an der FAU, Leiterin der Stammzellbiologischen Abteilung und Sprecherin des Zentrums für seltene Erkrankungen am Universitätsklinikum Erlangen. Ihr Labor erforscht, wodurch neurodegenerative Erkrankungen des Nervensystems, zu denen Motoneuronerkrankungen wie die ALS gehören, ausgelöst werden und welche Therapieansätze sich daraus ableiten lassen. „Seit etwa 15 Jahren ist bekannt, dass im Endstadium der ALS das Eiweiß TDP-43 in Neuronen unlöslich wird und verklumpt“, sagt Winner. „Es verliert dadurch seine normale Funktion und bekommt toxische Eigenschaften.“ Auch wenn diese pathologischen Veränderungen bei Patientinnen und Patienten en noch nicht zu erkennen sind, ist das Schicksal der Nervenzellen bereits entschieden. Winner: „Wir wollten wissen, ob wir Ursachen für die ALS in einem frühen Entwicklungsstadium nachweisen können, bevor sich TDP-43 verändert.“
Gemeinsam mit Prof. Dr. Jürgen Winkler und PD Dr. Martin Regensburger von der Abteilung für Molekulare Neurologie am Uni-Klinikum Erlangen ist Winner auf die Suche gegangen. Bedient haben die Forschenden sich dabei eines Kunstgriffs. Sie haben ALS-Patientinnen und -Patienten sowie gesunden Menschen einer Kontrollgruppe eine kleine Hautprobe aus dem Oberarm entnommen und in sogenannte induzierte pluripotente Stammzellen verwandelt – Zellen, die einem sehr frühen Entwicklungsstadium des Menschen gleichkommen und sich konzeptionell in jede beliebige Körperzelle entwickeln können. Diese Stammzellen wiederum wurden in Nervenzellen umgewandelt. „Wir haben sozusagen die Uhr zurückgedreht und Neurone generiert, die das Entwicklungsstadium eines Fötus nachahmen“, sagt Winner. Dass Körperzellen erwachsener Menschen in pluripotente Stammzellen zurück verwandelt werden können, wurde von Shin’ya Yamanaka entdeckt – 2012 erhielt der Japaner dafür den Medizin-Nobelpreis.
Das Eiweiß NOVA1 zeigt pathologische Merkmale bereits im Frühstadium
Mittels Massenspektrometrie, einem Hochdurchsatzverfahren, haben die Erlanger Forschenden die Zellproben nach unlöslichen Eiweißen durchsucht. Und sie wurden fündig: In den Nervenzellen der ALS-Patientinnen und -Patienten entdeckten sie ein RNA-bindendes Eiweiß mit dem Namen NOVA1. „Das Eiweiß zeigte eine deutlich erhöhte Unlöslichkeit und weitere Veränderungen in den Neuronen, aber noch nicht die klassischen TDP-43-Erkrankungsmerkmale“, erzählt Dr. Florian Krach, Mitglied des FAU-Teams und Erstautor der Studie. „Die Zellen der Kontrollgruppe wiesen diese Veränderungen nicht auf.“
Mit diesen Erkenntnissen wechselte Krach, gefördert vom Bayerisch-Kalifornischen Hochschulzentrum BaCaTeC, in das Labor des renommierten RNA-Biologen und Bioinformatikers Prof. Gene Yeo an der University of California in San Diego (USA). Dank spezialisierter Experimente und computergestützter Auswertung konnte er untersuchen, welche Bindungen NOVA1 in den RNA-Molekülen eingeht und welchen Einfluss es auf das alternative Spleißen in humanen Neuronen hat. „Das alternative Spleißen ist ein äußerst komplexer und ausgeklügelter Mechanismus, mit dem der Mensch sein Repertoire an Eiweißstoffen vervielfältigt“, erklärt Krach. „Dabei werden Abschnitte eines RNA-Botenmoleküls entweder herausgeschnitten oder hinzugefügt und auf diese Weise die Funktionen von Eiweißen gehemmt, erweitert oder gänzlich verändert.“
Die Forschenden hoffen auf frühe Diagnose und neue Therapiekonzepte
Dass der Prozess des alternativen Spleißens bei ALS-Patientinnen und -Patienten unreguliert abläuft, ist seit längerem bekannt. Bekannt war auch, dass TDP-43 diesen Prozess beeinflusst. Die Erlanger Forschenden hatten jedoch den Verdacht, dass in frühen Erkrankungsstadien, in denen TDP-43 noch nicht verändert ist, andere RNA-bindende Eiweiße für die pathologischen Vorgänge verantwortlich sind. Mit der Entdeckung der gestörten Funktion von NOVA1 hat sich dieser Verdacht nun bestätigt.
„Unsere Entdeckung ist bahnbrechend, aber nur ein erster Schritt hin zu einer möglichen Früherkennung von ALS“, sagt Beate Winner. „Folgende Studien mit größeren Kohorten könnten unsere Erkenntnisse über die Bedeutung von RNA-bindenden Proteinen vertiefen.“ Die Forscherinnen und Forscher hoffen, mit ihrer Arbeit dazu beitragen, neue Therapiekonzepte zu entwickeln – bevor Neurone einen Weg einschlagen, der keine Rückkehr erlaubt.
Weitere Informationen
DOI: https://doi.org/10.1007/s00401-022-02450-3
Prof. Dr. Beate Winner
Professur für Stammzellmodelle Seltener neuraler Erkrankungen
Tel.: 09131/85-39301
beate.winner@fau.de